Índice de contenidos
- 1 ¿Qué es el Angioedema Hereditario por déficit de C1 Inhibidor?
- 2 ¿En qué consiste el Angioedema Hereditario por déficit de C1 Inhibidor?
- 3 ¿Cuál es la visión del Angioedema Hereditario por déficit de C1 Inhibidor desde el punto de vista de las asociaciones de pacientes (FIGURA 1)?
- 4 ¿Cuál es la visión del Angioedema Hereditario por déficit de C1 Inhibidor desde el punto de vista de las sociedades médicas y/o científicas (5,10)?
- 5 ¿Cómo se tratan la mayoría de angioedemas “o hinchazones”? ¿Qué particularidad tiene el tratamiento del Angioedema Hereditario por déficit de C1 Inhibidor (AEH-C1-INH)?
- 6 ¿Cuál va a ser ese tratamiento?
- 7 ¿Cuáles son los factores que pueden precipitar un ataque de Angioedema Hereditario por déficit de C1 Inhibidor?
- 8 ¿Cómo se diagnostica el Angioedema Hereditario por Déficit de C1 Inhibidor?
- 9 ¿Cuál es el tratamiento en el AEH-C1INH?
- 10 Bibliografía
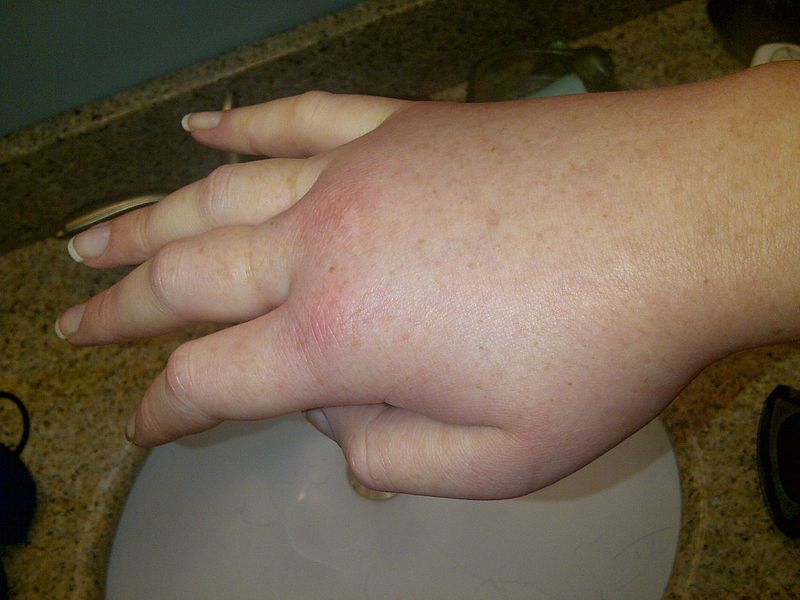
¿Qué es el Angioedema Hereditario por déficit de C1 Inhibidor?
{kind=link}
La prevalencia mínima de AEH en España es 1,09 por 100.000 habitantes, (1) pero al ser una enfermedad rara, algunos pacientes pueden estar mal diagnosticados y la prevalencia puede ser mayor. Hasta la fecha, ningún estudio ha demostrado diferencias en la prevalencia entre grupos étnicos.
¿En qué consiste el Angioedema Hereditario por déficit de C1 Inhibidor?
aparato digestivo, urinario y respiratorio. El AEH-C1INH es una enfermedad genética de carácter autosómico dominante caracterizada por una deficiencia de la proteína del Inhibidor de la C1 esterasa (C1 inhibidor) funcionalmente activa (6,7).
¿Cuál es la visión del Angioedema Hereditario por déficit de C1 Inhibidor desde el punto de vista de las asociaciones de pacientes (FIGURA 1)?
está formada por una red de asociaciones nacionales sin ánimo de lucro de pacientes con AEH.
.") |
|
Figura 1: Asociaciones de pacientes en relación con el Angioedema Hereditario mediado por Bradicinina (que incluye el déficit de C1 Inhibidor).
|
¿Cuál es la visión del Angioedema Hereditario por déficit de C1 Inhibidor desde el punto de vista de las sociedades médicas y/o científicas (5,10)?
a Grupo Español de Estudio del Angioedema mediado por Bradicinina (GEAB) (SGBA-Spanish Group for the study of Bradykinin induced Angioedema)(11).
siendo el último el Grupo de Trabajo Internacional sobre Angioedema Hereditario (del inglés: “Hereditary Angioedema International Working Group” – HAWK), que ha propuesto una clasificación para el AE mediado por BK12.
 |
|
Figura 2: Clasificación del angioedema según el Grupo de Trabajo
Internacional sobre Angioedema Hereditario13 |
¿Cómo se tratan la mayoría de angioedemas “o hinchazones”? ¿Qué particularidad tiene el tratamiento del Angioedema Hereditario por déficit de C1 Inhibidor (AEH-C1-INH)?
¿Cuál va a ser ese tratamiento?
mediadores de esta inflamación.
¿Cuáles son los factores que pueden precipitar un ataque de Angioedema Hereditario por déficit de C1 Inhibidor?
¿Cómo se diagnostica el Angioedema Hereditario por Déficit de C1 Inhibidor?
 |
|
FIGURA 1: Sospecha clínica del diagnóstico de AEH-C1-INH6
|
diagnóstico diferencial con abdomen agudo.
¿Cuál es el tratamiento en el AEH-C1INH?
(también llamado profilaxis a largo plazo), donde el paciente por lo general está tomando a diario un tratamiento oral, ya sean andrógenos atenuados (AA) o antifibrinolíticos (con sus respectivas contraindicaciones). Además, en algunos casos se utilizan preparados de terapia de reemplazo con pdhC1INH10.
después de cualquier procedimiento que conlleve un riesgo aumentado de desencadenar un ataque de AE. Por ello la importancia de la premedicación de cualquier procedimiento odontoestomatológico11, más que nada por la localización debido a la proximidad a la vía
aérea. También es importante la premedicación en cualquier procedimiento endoscópico u quirúrgico en general.
reemplazo con pdhC1INH (derivado de donaciones de sujetos sanos) y de bloqueantes de los receptores de Bradicinina BK14 (que al fin y al cabo es el mediador final común).
Bibliografía
1-Roche O, Blanch A, Caballero T, Sastre N, Callejo D, López-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94:498-503.
2- Peña J. Sistema inmune. Capítulo 1. Libro Inmunología (3ª edición, ISBN: 84-368-1213-1). Ed. Jose Peña. Editorial Pirámide (Grupo Anaya). 1998. pp.33-46.
3- García-Olivares E, Alonso A, Peña J. Complemento. Capítulo 13. Libro Inmunología (3ª edición, ISBN: 84-368-1213-1).Ed. Jose Peña. Editorial Pirámide (Grupo Anaya). 1998.pp. 225-238.
4- Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L et al. Hereditary and acquired angioedema: Problems and progress: Proceedings of the third C1 esterase Inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 2004; 114: S51-131.
5- Caballero T, Baeza ML, Cabañas R, Campos A, Cimbollek S, Gómez-Traseira C et al. Spanish consensus on the diagnosis, management and treatment of angio-oedema mediated by bradykinin. Part I. Classification, epidemiology, pathophysiology, genetics, clinical symptoms, and diagnosis. J Investig Allergol Clin Immunol. 2011 21: 333-47.
6-Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C1-esterase. Am J Med 1963; 31: 37-44.
7- Davis AE III, Whitehead AS, Harrison RA, Dauphinais A, Bruns GA, Cicardi M, et al. Human inhibitor of the first component of complement, C1: characterization of cDNA clones and localization of the gene to chromosome 11. Proc Natl Acad Sci USA 1986; 83:3161-5.
8- Asociación Española de Angioedema Familiar (por Deficiencia de C1 Inhibidor) (AEDAF)
9-Organización Internacional de Pacientes con Déficit de C1 Inhibidor (HAEI – International Patient Organization for C1-Inhibitor Deficiencies)
10-Caballero T, Baeza ML, Cabañas R, Campos A, Cimbollek S, Gómez-Traseira C et al. Spanish consensus on the diagnosis, management and treatment of angio-oedema mediated by bradykinin. Part II. Treatment, follow-up and special situations. J Investig Allergol Clin Immunol. 2011;21:422-41.
11- Grupo Español de Estudio del Angioedema mediado por Bradicinina” (GEAB) .
12- Cicardi M, Bork K, Caballero T, Craig T, Li HH, Longhurst H, Reshef A, Zuraw B; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67:147-57.
Hola tengo una deficit de c1 me gustaría saber más sobre ello , soy la primera de mi familia que la tiene me la diagnosticaron con 33 años ahora tengo 35 y por mucho que leo no me entero.